Laboratory of Dr. Ivy Dick
Calcium Channel Gating and Regualtion
Voltage-gated calcium channels (VGCCs) are critical conduits for calcium entry into excitable cells and play essential roles in numerous physiological functions including muscle contraction, neurotransmission, gene transcription, and cardiac rhythm. As such, calcium entry through these channels is precisely controlled through both calcium and voltage dependent feedback mechanisms. The calcium dependent regulation of these channels is controlled by the calcium sensor calmodulin, which resides on the carboxy-terminus of the channel and tunes the response of the channel for distinct physiological functions across a variety of cell types. Such calcium dependent regulation can be modulated by numerous mechanisms including alternative splicing of the channel, RNA editing, and protein binding. Our lab probes the mechanisms underlying such modulation, and has identified key components within the channel which underly context specific changes in channel regulation.
Calcium Channelopathies
Voltage-gated calcium channels are critical conduits for calcium entry into the heart, smooth muscle and brain. Disruption of the normal gating and regulation of these channels results in severe clinical phenotypes including autism, ataxia and cardiac arrhythmia. Our lab has focused on gaining mechanistic understanding of how distinct channel mutations alter the biophysical properties of the channel, with the aim of gaining new insight into the pathogenesis and treatment options for calcium channelopathies and related diseases.
Cardiac Arrythmia: The CaV1.2 L-type calcium channel is critical for normal cardiac function. These channels are open during the plateau phase of the cardiac action potential, and are thus important determinants of the duration of the action potential. Mutations which reduce the calcium dependent inactivation of these channels cause a substantial prolongation of the action potential duration, corresponding to severe long-QT syndrome in patients. Calmodulinopathies represent one such case, where we have shown that a mutation in calmodulin can cause a loss of calcium dependent inactivation of CaV1.2. Evaluation of the action potential through current clamp recordings of a ventricular myocyte harboring a calmodulinopathy mutation demonstrates marked action potential prolongation, indicative of long-QT syndrome. Alternatively, mutations directly on the CaV1.2 channel cause a disease known as Timothy syndrome or Long-QT type 8. Our lab has demonstrated that these mutations can exert a variety of effects on channel gating, with distinct effects on voltage and calcium dependent inactivation. This deficit in channel inactivation results in long-QT syndrome with episodes of life-threatening cardiac arrhythmia. In order to study the pathogenesis of these mutations, we have generated induced pluripotent stem cell (iPSC) derived cardiomyocytes harboring select Timothy syndrome mutations via CRISPR editing. Optical mapping of the iPSC derived cardiomyocytes reveals a spiral wave pattern in cells harboring the mutation, indicative of a reentrant arrhythmia. Using a combination of patch clamp recordings and imaging, we can probe the pathogenic mechanisms underlying these cardiac phenotypes, with the goal of providing new insight into the pathogenesis and treatment of calcium channelopathies. Our ongoing research includes the development of a novel treatment for CaV1.2 channelopathies, demonstrating how in-depth biophysics can guide new therapeutic avenues.
Typical Cardiac Action Potential
current clamp recording of a ventricular myocyte

Calmodulinopathy Mutation
current clamp recording of a ventricular myocyte
Action Potential Propagation in Cardiomyocytes
optical mapping of iPSC derived cardiomyocytes

Timothy Syndrome Produces Arrythmia
optical mapping recording across a monolayer of iPSC derived cardiomyocytes harboring a TS mutation

Enhanced Activation Associates with ASD and neurodevelopmental delay
cartoon depicting the impact of S6 mutations on channel function and patient phenotype
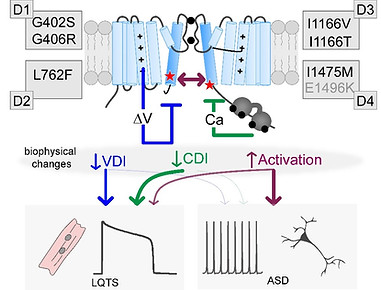
Neurological Disease: Voltage-gated calcium channels are critical for normal neurological function and are known to play a major role in neuronal excitation, transcription and long-term memory. CaV2.1 channels are required for synaptic transmission, and mutations that alter the function of these channels are causative of multiple phenotypes including ataxia, migraine and epilepsy. In addition, L-type calcium channels are highly associated with Biopolar Disorder and Schizophrenia. Moreover, many of the same mutations in CaV1.2 which cause Long-QT syndrome also cause neurodevelopmental delay and autism spectrum disorder in patients. Our lab has focused on understanding the impact of these mutations on channel gating. Our results thus far may provide a mechanistic explanation for the neurodevelopmental effects of select mutations, positioning a hyperpolerizing shift in channel activation as a critical feature underlying the neurological effects of TS mutations.
Contact Us
Thanks for your interest in our research. Get in touch with us for any questions or comments.
University of Maryland School of Medicine
660 W. Redwood Street
Howard Hall Room 511
